Investigations into Cancer–Immune System Interactions
Immune cells can infiltrate the tumor microenvironment, the consequences of which are not well understood. The multi-faceted immune presence interacts with cancer cells in inhibitory and/or stimulatory ways, resulting in complex cancer–immune interactions. Below we describe our current mathematical and computational approaches to understand the consequences and implications of these intercellular interactions.
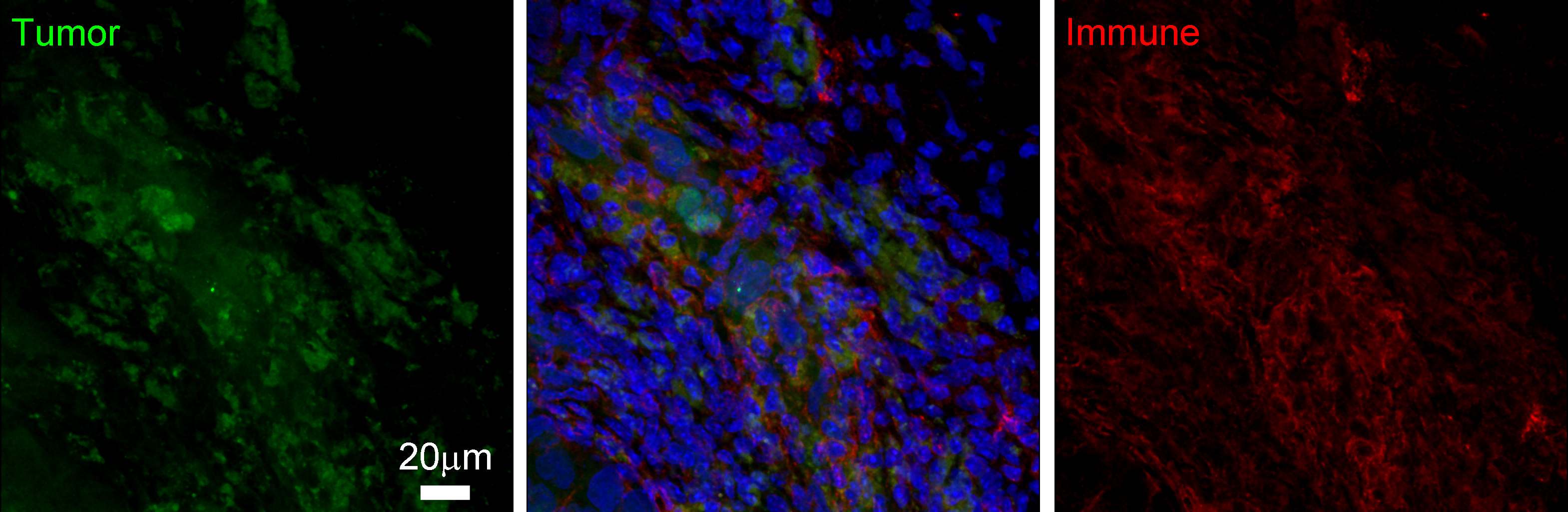
Tumor-Promoting Inflammation
The presence of cancer within a host initiates a systemic immune
response towards the transformed cells. Inflammatory immune cells
such as neutrophils, platelets, macrophages, and natural killer
cells, are recruited to the tumor site where they initiate the wound
healing process. Tumors, sometimes viewed as wounds that never heal,
can be promoted by these inflammatory actions. 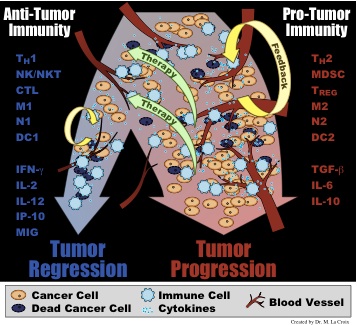 Once the adaptive
immune response is activated by dendritic cells and macrophages,
CD8+ T cells, or cytotoxic T lymphocytes, infiltrate the tumor and
induce apoptosis in the target tumor cells. Depending on the
cytokines and other signals present in the tumor microenvironment,
recruited immune cells will either form a pro-tumor immunity
(typified by cytokines such as TGF-β, IL-6, and IL-10 and cells
such as M2 macrophages, Th-2 T helper cells, and myeloid derived
suppressor cells) or an anti-tumor immunity (typified by cytokines
such as IFN-γ, IL-2, and IL-12 and cells such as M1
macrophages, Th-1 T helper cells, and cytotoxic lymphocytes).
Once the adaptive
immune response is activated by dendritic cells and macrophages,
CD8+ T cells, or cytotoxic T lymphocytes, infiltrate the tumor and
induce apoptosis in the target tumor cells. Depending on the
cytokines and other signals present in the tumor microenvironment,
recruited immune cells will either form a pro-tumor immunity
(typified by cytokines such as TGF-β, IL-6, and IL-10 and cells
such as M2 macrophages, Th-2 T helper cells, and myeloid derived
suppressor cells) or an anti-tumor immunity (typified by cytokines
such as IFN-γ, IL-2, and IL-12 and cells such as M1
macrophages, Th-1 T helper cells, and cytotoxic lymphocytes).
To investigate the role of tumor-promoting inflammation, an emerging
hallmark of cancer, we have developed a mathematical model for
cancer-immune interactions that can capture both the pro-angiogenic,
tumor-progressing actions of a pro-tumor inflammatory
microenvironment, and the anti-angiogenic, tumor-inhibiting actions
of an anti-tumor inflammatory microenvironment. This model utilizes
principles of generalized logistic growth, which captures some of
the inherent variability underlying tumor growth in an immune
competent host that is often neglected in macroscopic measurements
and in mathematical models. From model simulations, the two types of
inflammation (pro-tumor or anti-tumor) resolve into two
fundamentally different classes of outcomes, where
inflammation-enhanced tumor progression must either result in a
decreased tumor burden, as in the anti-tumor case, or in an
increased tumor burden, as in the pro-tumor case. These results
suggest that, in some cases, fast tumor growth may be advantageous,
if it leads to a significantly smaller tumor burden. In such cases,
it is possible that treatments should be targeted towards enhancing
the stability of an anti-tumor inflammatory environment instead of
towards immediate tumor regression.
Tumor Immunoevasion
Despite highly evolved adaptive immune responses, tumors often manage to escape recognition by the immune system. This process is known as immunoevasion, and is another emerging hallmark of cancer.
When hematopoietic stem cells leave the bone marrow they differentiate into either lymphoid progenitors or myeloid progenitors. Lymphoid progenitors migrate to the thymus where they differentiate into T, B, and NKT cells. Myeloid progenitors differentiate into monocytes, migrate to tissues, and differentiate into myeloid cells such as dendritic cells (DCs) and macrophages. When an immature DC encounters an antigen, it internalizes the antigen to display fragments on its membrane. The DC then matures as it migrates to a lymph node. Maturation involves the loss of ability to engulf pathogens and an increased ability to communicate with T cells. Within the lymph nodes (collection points where antigen presenting cells interact with T cells attracted to the node via chemotaxis), mature DCs activate naïve T cells to develop a specific immune response. Activated cytotoxic T cells undergo rapid clonal expansion and migrate throughout the body in search of relevant targets. T cells perform their cytotoxic function by inducing apoptosis in the target cell through the secretion of perforin and granzymes or through Fas/Fas-ligand binding.
Within the process of activating the adaptive immune response
described above, two significant functions may be subverted by tumors:
antigen presentation (maturation of DCs) and T cell functionality.
Antigen presentation suppression
If antigen presentation is blocked then naïve T cells are not activated and a specific immune response is not mounted. A number of cytokines, chemokines and growth factors, such as HIF-1α, VEGF, nitric oxide, and reactive oxygen species (ROS), produced within the tumor microenvironment may interfere with the process of DC maturation. Without DC maturation, there may be an accumulation of immunosuppressive factors, such as myeloid derived suppressor cells (MDSCs), in the tumor microenvironment resulting in immunoevasion.
In order to investigate this process from a modeling perspective, we
formulated a system of ordinary differential equations in a
predator-prey type system, where the prey (cancer cells) have a
defense mechanism (immunoevasion) against recognition by the predator
(immune system). Our analysis suggests that this mechanism can have
significant effects on overall tumor-immune dynamics, ultimately
allowing for either tumor suppression or tumor escape in a manner that
depends on the strength of the immune suppression [Kareva et al,
2010]. Currently, we are investigating the possible role of glycolysis
and the resulting reduction of pH in a hypoxic tumor microenvironment
as another possible mechanism for immune evasion.
Impaired T cell functionality and immune resistance
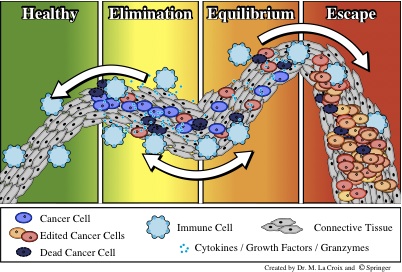 The immune response poses a second barrier to tumor growth after the
angiogenic switch. Immune surveillance of tissues allows for early
detection of transformed cells. If these transformed cells are not
recognized as “self”, they are eliminated by the immune
system. Through repeated exposure of the transformed cells to this
immune selection process, various phenotypes can arise within the
cancer cell population, creating a heterogeneous population of
neoplastic cells. These immunoedited cells may develop the ability to
evade the immune response and grow in an uncontrolled manner.
The immune response poses a second barrier to tumor growth after the
angiogenic switch. Immune surveillance of tissues allows for early
detection of transformed cells. If these transformed cells are not
recognized as “self”, they are eliminated by the immune
system. Through repeated exposure of the transformed cells to this
immune selection process, various phenotypes can arise within the
cancer cell population, creating a heterogeneous population of
neoplastic cells. These immunoedited cells may develop the ability to
evade the immune response and grow in an uncontrolled manner.
After prolonged periods of immune-induced dormancy, T cells can lose effectiveness in their cytotoxicity. This loss may be due to either T cell tolerization to the cancer cells or to an increased cancer cell resistance to immune attack. Both of these mechanisms are intertwined in the process of immunoediting that can lead to tumor escape from immune control.
To investigate the heterogeneous population-level dynamics involved in
this immune selection process, we are working on a mathematical model
that can capture the essential cancer-immune interactions that may
lead to T cell tolerization and / or the accumulation of
immune-resistance by cancer cells. These two fundamentally different
mechanisms of immune evasion would require specifically targeted
therapies, which could be analyzed theoretically with this
mathematical model.
Cancer Stem Cells and Immune System-Modulated Tumor Progression
The role of the immune system in tumor progression has been subject to discussion for many decades. Numerous studies suggest that a low immune response might be beneficial, if not necessary, for tumor growth, and only a strong immune response can counter tumor growth and thus inhibit progression.
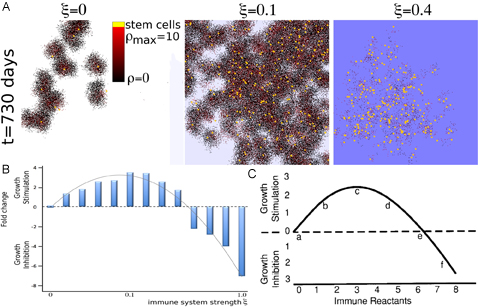 Without an immune response, a heterogeneous tumor population comprised
of cancer stem cells and non-stem progenitors grows as conglomerates
of self-metastases [Enderling et al., 2009]. This morphological
phenomenon results from the interplay of cell proliferation, cell
migration and cell death. With increasing cell death intra-tumoral
spatial inhibitions are loosened, which in turn enable cancer stem
cell cycling and thus, counter-intuitively, tumor progression [Enderling et al., 2009b]. By overlaying on this
model the diffusion of immune reactants into the tumor from a
peripheral source to target cells, we simulate the process of
immune-system-induced cell kill on tumor progression. A low cytotoxic
immune reaction continuously kills cancer cells and, although at a low
rate, thereby causes the liberation of space-constrained cancer stem
cells to drive self-metastatic progression and continued tumor
growth. With increasing immune system strength, however, tumor growth
peaks, and then eventually falls below the intrinsic tumor sizes
observed without an immune response. Focusing only on the cytotoxic
function of the immune system, we were able to observe all
immunoediting roles of the immune system: immune promotion at weak
immune responses, immunoinhibition at strong immune responses, and
immunoselection at all levels. Simulations of our model support a
hypothesis previously put forward by Prehn [Prehn, 1972] that comparable tumor sizes can be
observed for weak and strong immune reactions. With this increasing
immune response the number and proportion of cancer stem cells
monotonically increases, implicating an additional unexpected
consequence, that of cancer stem cell selection, to the immune
response.
Without an immune response, a heterogeneous tumor population comprised
of cancer stem cells and non-stem progenitors grows as conglomerates
of self-metastases [Enderling et al., 2009]. This morphological
phenomenon results from the interplay of cell proliferation, cell
migration and cell death. With increasing cell death intra-tumoral
spatial inhibitions are loosened, which in turn enable cancer stem
cell cycling and thus, counter-intuitively, tumor progression [Enderling et al., 2009b]. By overlaying on this
model the diffusion of immune reactants into the tumor from a
peripheral source to target cells, we simulate the process of
immune-system-induced cell kill on tumor progression. A low cytotoxic
immune reaction continuously kills cancer cells and, although at a low
rate, thereby causes the liberation of space-constrained cancer stem
cells to drive self-metastatic progression and continued tumor
growth. With increasing immune system strength, however, tumor growth
peaks, and then eventually falls below the intrinsic tumor sizes
observed without an immune response. Focusing only on the cytotoxic
function of the immune system, we were able to observe all
immunoediting roles of the immune system: immune promotion at weak
immune responses, immunoinhibition at strong immune responses, and
immunoselection at all levels. Simulations of our model support a
hypothesis previously put forward by Prehn [Prehn, 1972] that comparable tumor sizes can be
observed for weak and strong immune reactions. With this increasing
immune response the number and proportion of cancer stem cells
monotonically increases, implicating an additional unexpected
consequence, that of cancer stem cell selection, to the immune
response.
Cancer stem cells and immune cytotoxicity alone are
sufficient to explain the three-step “immunoediting”
concept — the modulation of tumor growth through inhibition,
selection, and promotion. We propose more generally that a
stem-cell-expansive influence may take the form of anything that
encourages morphological fingering. Beyond immune response, this could
include cell death, or even growth within restricted thin channels, as
might be expected e.g. during invasion of host tissue.
Resources
Publications:
A strong body of work in tumor immunology has been published by
researchers at CCSB (click on title to go to manuscript abstract):
- Beheshti A, Wage J, McDonald JT, Lamont
C, Peluso M, Hahnfeldt P, Hlatky L.
Tumor-host signaling interaction reveals a systemic, age-dependent splenic immune influence on tumor development.
Oncotarget. 2015 Nov 3;6(34):35419-32. [Open Access]
- Wage J, Ma L, Peluso M, Lamont C,
Evens AM, Hahnfeldt P, Hlatky L, Beheshti A.
Proton irradiation impacts age-driven modulations of cancer
progression influenced by immune system transcriptome modifications
from splenic tissue. J
Radiat Res. 2015 Sep;56(5):792-803. Epub 2015 Aug 7. PMCID: PMC4577010 [Open
Access]
- Kareva I, Wilkie KP, Hahnfeldt P. The Power of the Tumor Microenvironment: A Systemic
Approach for a Systemic Disease. In: d'Onofrio A, Gandolfi A
(eds). Mathematical Oncology 2013. Birkhäuser Basel,
2014:181-196. In Series: Modeling and Simulation in Science,
Engineering and Technology: Bellomo N (series ed).
- Wilkie KP, Hahnfeldt P. Mathematical models of
immune-induced cancer dormancy and the emergence of immune
evasion. Interface Focus. 2013 Aug 6;3(4):20130010. Epub 2013 Jun
25. PMCID: PMC3915830 [Open Access]
- Pilatova K, Greplova K, Demlova R, Bencsikova B, Klement
GL, Zdrazilova-Dubska L. Role of platelet chemokines, PF-4 and
CTAP-III, in cancer biology. J Hematol Oncol. 2013 Jun 24;6:42. PMCID: PMC3694472 [Open
Access]
- Wilkie KP, Hahnfeldt P. Tumor–immune dynamics regulated in the
microenvironment inform the transient nature of immune-induced tumor
dormancy. Cancer Res. 2013 Jun 15;73(12):3534-44. Epub 2013 Mar
27. PMCID: PMC3955200 [Open Access]
- Wilkie KP, Hahnfeldt P. Modeling the Dichotomy
of the Immune Response to Cancer: Cytotoxic Effects and
Tumor-Promoting Inflammation. arXiv:1305.3634 [q-bio.CB]. 2013 May
15.
- Kareva I, Hahnfeldt P. The emerging
“hallmarks” of metabolic
reprogramming and immune evasion: distinct or linked? Cancer
Res. 2013 May 1;73(9):2737-42. Epub 2013 Feb 19. [Open Access]
- Hillen T, Enderling H, Hahnfeldt P. The tumor
growth paradox and immune system-mediated selection for cancer stem
cells. Bull Math Biol. 2013 Jan;75(1):161-84. Epub 2012 Nov 30.
- Wilkie KP. A review of mathematical models of
cancer-immune interactions in the context of tumor dormancy. Adv Exp
Med Biol. 2013;734:201-34. (also in Systems Biology of Tumor Dormancy. Enderling H,
Almog N, Hlatky L, editors. New York: Springer, 2013; 201-34.)
- Enderling H, Hlatky L, Hahnfeldt P. Immunoediting: evidence of the multifaceted role of
the immune system in self-metastatic tumor growth. Theor Biol
Med Model. 2012 Jul 28;9(1):31. Epub 2012 Jul 28. PMCID: PMC3499182 [Open Access]
- Domhan S, Zeier M, Abdollahi A. Immunosuppressive therapy
and post-transplant malignancy. Nephrol Dial Transplant. 2009
Apr;24(4):1097-103. Epub 2008 Oct 31. [Open Access]